Fibrose cística

A fibrose cística é uma doença que causa o acúmulo de muco espesso e pegajoso nos pulmões, no trato digestivo e em outras áreas do corpo. É uma das doenças pulmonares crônicas mais comuns em crianças e adultos jovens. É um distúrbio com risco de vida.
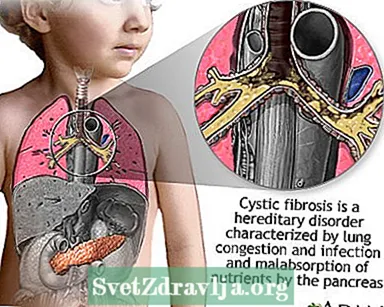
A fibrose cística (FC) é uma doença transmitida de família. É causada por um gene defeituoso que faz com que o corpo produza um fluido anormalmente espesso e pegajoso, chamado muco. Esse muco se acumula nas vias respiratórias dos pulmões e no pâncreas.
O acúmulo de muco resulta em infecções pulmonares com risco de vida e sérios problemas de digestão. A doença também pode afetar as glândulas sudoríparas e o sistema reprodutivo do homem.
Muitas pessoas são portadoras do gene da FC, mas não apresentam sintomas. Isso ocorre porque uma pessoa com FC deve herdar 2 genes defeituosos, um de cada pai. Alguns americanos têm o gene CF. É mais comum entre os descendentes do norte ou centro europeu.
A maioria das crianças com FC é diagnosticada aos 2 anos, especialmente porque a triagem neonatal é realizada nos Estados Unidos. Para um pequeno número, a doença não é detectada até os 18 anos ou mais. Essas crianças costumam ter uma forma mais branda da doença.
Os sintomas em recém-nascidos podem incluir:
- Crescimento atrasado
- Falha em ganhar peso normalmente durante a infância
- Sem movimentos intestinais nas primeiras 24 a 48 horas de vida
- Pele com gosto salgado
Os sintomas relacionados à função intestinal podem incluir:
- Dor de barriga devido a constipação intensa
- Aumento de gases, inchaço ou barriga que parece inchada (distendida)
- Náusea e perda de apetite
- Fezes que são claras ou cor de argila, cheirosas, com muco ou que flutuam
- Perda de peso
Os sintomas relacionados aos pulmões e seios da face podem incluir:
- Tosse ou aumento de muco nos seios da face ou nos pulmões
- Fadiga
- Congestão nasal causada por pólipos nasais
- Episódios repetidos de pneumonia (os sintomas de pneumonia em alguém com fibrose cística incluem febre, aumento da tosse e falta de ar, aumento do muco e perda de apetite)
- Dor ou pressão nos seios da face causada por infecção ou pólipos
Sintomas que podem ser observados mais tarde na vida:
- Infertilidade (em homens)
- Inflamação repetida do pâncreas (pancreatite)
- Sintomas respiratórios
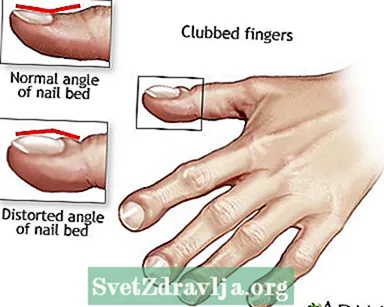
- Dedos machucados
Um exame de sangue é feito para ajudar a detectar a FC. O teste procura mudanças no gene CF. Outros testes usados para diagnosticar FC incluem:
- O teste de tripsinogênio imunorreativo (IRT) é um teste padrão de triagem neonatal para FC. Um alto nível de IRT sugere possível CF e requer mais testes.
- O teste de cloreto de suor é o teste de diagnóstico padrão para FC. Um alto nível de sal no suor da pessoa é um sinal da doença.
Outros testes que identificam problemas que podem estar relacionados à CF incluem:
- Radiografia de tórax ou tomografia computadorizada
- Teste de gordura fecal
- Testes de função pulmonar
- Medição da função pancreática (elastase pancreática nas fezes)
- Teste de estimulação de secretina
- Tripsina e quimiotripsina nas fezes
- Série GI superior e intestino delgado
- Culturas pulmonares (obtidas por escarro, broncoscopia ou esfregaço de garganta)
Um diagnóstico precoce de FC e um plano de tratamento podem melhorar a sobrevida e a qualidade de vida. Acompanhamento e monitoramento são muito importantes. Quando possível, o atendimento deve ser recebido em uma clínica especializada em fibrose cística. Quando as crianças atingem a idade adulta, devem ser transferidas para um centro especializado em fibrose cística para adultos.
O tratamento para problemas pulmonares inclui:
- Antibióticos para prevenir e tratar infecções pulmonares e sinusais. Eles podem ser tomados por via oral, ou administrados nas veias ou por tratamentos respiratórios. Pessoas com FC podem tomar antibióticos apenas quando necessário ou o tempo todo. As doses costumam ser maiores do que o normal.
- Medicamentos inalados para ajudar a abrir as vias respiratórias.
- Outros medicamentos administrados por meio de um tratamento respiratório para diluir o muco e torná-lo mais fácil de tossir são a terapia com enzimas DNAse e soluções salinas altamente concentradas (solução salina hipertônica).
- A vacina contra a gripe e a vacina pneumocócica polissacarídica (PPV) anualmente (pergunte ao seu médico).
- O transplante de pulmão é uma opção em alguns casos.
- A oxigenoterapia pode ser necessária à medida que a doença pulmonar piora.
Os problemas pulmonares também são tratados com terapias para diluir o muco. Isso torna mais fácil tossir o muco dos pulmões.
Esses métodos incluem:
- Atividade ou exercício que o faz respirar profundamente
- Dispositivos que são usados durante o dia para ajudar a limpar as vias respiratórias de muito muco
- Percussão torácica manual (ou fisioterapia torácica), na qual um membro da família ou um terapeuta bate levemente no peito, nas costas e na área sob os braços da pessoa
O tratamento para problemas nutricionais e intestinais pode incluir:
- Uma dieta especial rica em proteínas e calorias para crianças mais velhas e adultos
- Enzimas pancreáticas para ajudar a absorver gorduras e proteínas, que são tomadas com todas as refeições
- Suplementos vitamínicos, especialmente vitaminas A, D, E e K
- Seu provedor pode recomendar outros tratamentos se você tiver fezes muito duras
Ivacaftor, lumacaftor, tezacaftor e elexacaftor são medicamentos que tratam certos tipos de FC.
- Eles melhoram a função de um dos genes defeituosos que causam a FC.
- Até 90% dos pacientes com FC e elegíveis para um ou mais desses medicamentos isoladamente ou em combinação.
- Como resultado, há menos acúmulo de muco espesso nos pulmões. Outros sintomas de FC também melhoraram.
Os cuidados e monitoramento em casa devem incluir:
- Evitar fumaça, poeira, sujeira, fumos, produtos químicos domésticos, fumaça de lareira e mofo ou bolor.
- Dar muitos líquidos, especialmente para bebês e crianças em climas quentes, quando houver diarreia ou fezes moles, ou durante atividades físicas extras.
- Praticar exercícios 2 ou 3 vezes por semana. Natação, corrida e ciclismo são boas opções.
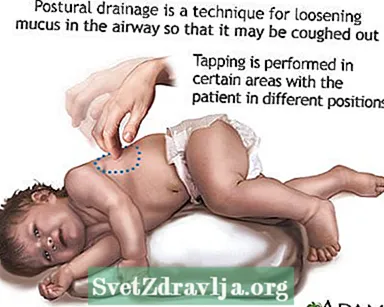
- Limpar ou expelir muco ou secreções das vias aéreas. Isso deve ser feito 1 a 4 vezes por dia. Pacientes, familiares e cuidadores devem aprender a fazer percussão torácica e drenagem postural para ajudar a manter as vias aéreas desobstruídas.
- Nenhum contato com outras pessoas com FC é recomendado, pois eles podem trocar infecções (não se aplica a membros da família).
Você pode aliviar o estresse da doença ingressando em um grupo de apoio à fibrose cística. Compartilhar com outras pessoas que tenham experiências e problemas comuns pode ajudar sua família a não se sentir sozinha.
A maioria das crianças com FC permanece com boa saúde até atingir a idade adulta. Eles podem participar da maioria das atividades e frequentar a escola. Muitos jovens adultos com FC terminam a faculdade ou encontram empregos.
A doença pulmonar eventualmente piora a ponto de a pessoa ficar incapacitada. Hoje, a expectativa de vida média para pessoas com FC que chegam à idade adulta é de cerca de 44 anos.
A morte é geralmente causada por complicações pulmonares.
A complicação mais comum é a infecção respiratória crônica.
Outras complicações incluem:
- Problemas intestinais, como cálculos biliares, bloqueio intestinal e prolapso retal
- Tossindo sangue
- Insuficiência respiratória crônica
- Diabetes
- Infertilidade
- Doença hepática ou insuficiência hepática, pancreatite, cirrose biliar
- Desnutrição
- Pólipos nasais e sinusite
- Osteoporose e artrite
- Pneumonia que não para de voltar
- Pneumotórax
- Insuficiência cardíaca direita (cor pulmonale)
- Câncer colorretal
Ligue para o seu provedor se um bebê ou criança apresentar sintomas de FC e experimentar:
- Febre, aumento da tosse, alterações na expectoração ou sangue na expectoração, perda de apetite ou outros sinais de pneumonia
- Maior perda de peso
- Evacuações intestinais ou fezes mais frequentes que cheiram mal ou têm mais muco
- Barriga inchada ou aumento do inchaço
Ligue para o seu médico se uma pessoa com FC desenvolver novos sintomas ou se os sintomas piorarem, especialmente dificuldade respiratória grave ou tosse com sangue.
CF não pode ser evitado. O rastreamento de pessoas com histórico familiar da doença pode detectar o gene da FC em muitos portadores.
CF
- Nutrição enteral - criança - gerenciamento de problemas
- Tubo de alimentação de gastrostomia - bolo
- Como respirar quando você está com falta de ar
- Tubo de alimentação de jejunostomia
- Drenagem postural
 Clubbing
Clubbing Drenagem postural
Drenagem postural Dedos machucados
Dedos machucados Fibrose cística
Fibrose cística
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor / ivacaftor em indivíduos com fibrose cística e F508del / F508del-CFTR ou F508del / G551D-CFTR. Am J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Fibrose cística. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21ª ed. Filadélfia, PA: Elsevier; 2020: cap 432.
Farrell PM, White TB, Ren CL, et al. Diagnóstico de fibrose cística: diretrizes consensuais da Cystic Fibrosis Foundation. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Efeitos da terapia com lumacaftor / ivacaftor na função CFTR em pacientes homozigotos Phe508del com fibrose cística. Am J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Cystic fibrosis. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 26ª ed. Filadélfia, PA: Elsevier; 2020: cap 83.
Rowe SM, Hoover W., Solomon GM, Sorscher EJ. Fibrose cística. Em: Broaddus VC, Mason RJ, Ernst JD, et al, eds. Livro de Murray e Nadel de Medicina Respiratória. 6ª ed. Filadélfia, PA: Elsevier Saunders; 2016: cap 47.
Taylor-Cousar JL, Munck A., McKone EF, et al. Tezacaftor-ivacaftor em pacientes com fibrose cística homozigótica para phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Interessante

Tratar minha disfunção erétil salvou minha vida
