Janela aortopulmonar

A janela aortopulmonar é um defeito cardíaco raro no qual há um orifício que conecta a artéria principal que leva o sangue do coração para o corpo (a aorta) e a que leva o sangue do coração para os pulmões (artéria pulmonar). A condição é congênita, o que significa que está presente no nascimento.
Normalmente, o sangue flui através da artéria pulmonar para os pulmões, onde recebe oxigênio. Em seguida, o sangue retorna ao coração e é bombeado para a aorta e o resto do corpo.
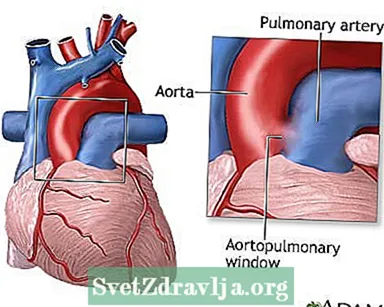
Bebês com janela aortopulmonar apresentam um orifício entre a aorta e a artéria pulmonar. Por causa desse orifício, o sangue da aorta flui para a artéria pulmonar e, como resultado, muito sangue flui para os pulmões. Isso causa pressão alta nos pulmões (uma condição chamada hipertensão pulmonar) e insuficiência cardíaca congestiva. Quanto maior o defeito, mais sangue consegue entrar na artéria pulmonar.
A condição ocorre quando a aorta e a artéria pulmonar não se dividem normalmente à medida que o bebê se desenvolve no útero.
A janela aortopulmonar é muito rara. É responsável por menos de 1% de todos os defeitos cardíacos congênitos.
Esta condição pode ocorrer por si só ou com outros defeitos cardíacos, como:
- Tetralogia de Fallot
- Atresia pulmonar
- Truncus arteriosus
- Defeito do septo atrial
- Persistência do canal arterial
- Arco aórtico interrompido
Cinquenta por cento das pessoas geralmente não têm outros defeitos cardíacos.
Se o defeito for pequeno, pode não causar nenhum sintoma. No entanto, a maioria dos defeitos são grandes.
Os sintomas podem incluir:
- Crescimento atrasado
- Insuficiência cardíaca
- Irritabilidade
- Má alimentação e falta de ganho de peso
- Respiração rápida
- Batimento cardíaco acelerado
- Infecções respiratórias
O médico geralmente ouvirá um som cardíaco anormal (sopro) ao ouvir o coração da criança com um estetoscópio.
O provedor pode solicitar testes como:
- Cateterismo cardíaco - um tubo fino inserido nos vasos sanguíneos e / ou artérias ao redor do coração para visualizar o coração e os vasos sanguíneos e medir diretamente a pressão no coração e nos pulmões.
- Raio-x do tórax.
- Ecocardiograma.
- Ressonância magnética do coração.
A condição geralmente requer cirurgia de coração aberto para reparar o defeito. A cirurgia deve ser feita o mais rápido possível após o diagnóstico. Na maioria dos casos, isso ocorre quando a criança ainda é um recém-nascido.
Durante o procedimento, uma máquina coração-pulmão assume o controle do coração da criança. O cirurgião abre a aorta e fecha o defeito com um remendo feito de um pedaço do saco que envolve o coração (o pericárdio) ou de um material artificial.
A cirurgia para corrigir a janela aortopulmonar é bem-sucedida na maioria dos casos. Se o defeito for tratado rapidamente, a criança não deve ter nenhum efeito duradouro.
Atrasar o tratamento pode levar a complicações como:
- Insuficiência cardíaca congestiva
- Hipertensão pulmonar ou síndrome de Eisenmenger
- Morte
Ligue para o seu provedor se seu filho tiver sintomas de janela aortopulmonar. Quanto mais cedo essa condição for diagnosticada e tratada, melhor será o prognóstico da criança.
Não há maneira conhecida de prevenir a janela aortopulmonar.
Defeito do septo aortopulmonar; Fenestração aortopulmonar; Defeito cardíaco congênito - janela aortopulmonar; Coração com defeito congênito - janela aortopulmonar
 Janela aortopulmonar
Janela aortopulmonar
Fraser CD, Kane LC. Doença cardíaca congênita. In: Townsend CM, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery. 20ª ed. Filadélfia, PA: Elsevier; 2017: cap 58.
Qureshi AM, Gowda ST, Justino H, Spicer DE, Anderson RH. Outras malformações das vias de saída ventricular. Em: Wernovsky G, Anderson RH, Kumar K, et al, eds. Cardiologia Pediátrica de Anderson. 4ª ed. Filadélfia, PA: Elsevier; 2020: cap 51.
Webb GD, Smallhorn JF, Therrien J, Redington AN. Cardiopatia congênita no paciente adulto e pediátrico. In: Zipes DP, Libby P, Bonow RO, Mann DL, Tomaselli GF, Braunwald E, eds. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 11ª ed. Filadélfia, PA: Elsevier; 2019: cap 75.
Artigos Fascinantes

Síndrome do leite-álcali
